PTT
La púrpura trombocitopénica trombótica (PTT) es un síndrome clínico 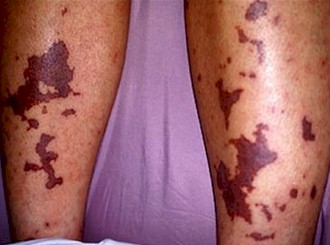 caracterizado por anemia hemolítica microangiopática, trombocitopenia, fiebre, déficits neurológicos fluctuantes y enfermedad renal. Su asociación con lupus eritematoso sistémico (LES) se reconoció en la literatura médica desde 1939. Las dos enfermedades presentan manifestaciones clínicas simmilares por lo que en ocasiones es dificultoso establecer la presencia de PTT en un paciente con LES activo. La identificación de esquistocitos en el frotis de sangre periférica es crucial para el diagnóstico, así como la reticulocitosis marcada y la negatividad en la reacción de Coombs directa. Presentamos tres pacientes de sexo feminino en las que las dos entidades se presentaron en forma simultánea. Sugerimos la utilización de inmunosupresores junto con tratamiento de plasmaféresis con reposición de sobrenadante de crioprecipitado. (AU)
caracterizado por anemia hemolítica microangiopática, trombocitopenia, fiebre, déficits neurológicos fluctuantes y enfermedad renal. Su asociación con lupus eritematoso sistémico (LES) se reconoció en la literatura médica desde 1939. Las dos enfermedades presentan manifestaciones clínicas simmilares por lo que en ocasiones es dificultoso establecer la presencia de PTT en un paciente con LES activo. La identificación de esquistocitos en el frotis de sangre periférica es crucial para el diagnóstico, así como la reticulocitosis marcada y la negatividad en la reacción de Coombs directa. Presentamos tres pacientes de sexo feminino en las que las dos entidades se presentaron en forma simultánea. Sugerimos la utilización de inmunosupresores junto con tratamiento de plasmaféresis con reposición de sobrenadante de crioprecipitado. (AU)
caracterizado por anemia hemolítica microangiopática, trombocitopenia, fiebre, déficits neurológicos fluctuantes y enfermedad renal. Su asociación con lupus eritematoso sistémico (LES) se reconoció en la literatura médica desde 1939. Las dos enfermedades presentan manifestaciones clínicas simmilares por lo que en ocasiones es dificultoso establecer la presencia de PTT en un paciente con LES activo. La identificación de esquistocitos en el frotis de sangre periférica es crucial para el diagnóstico, así como la reticulocitosis marcada y la negatividad en la reacción de Coombs directa. Presentamos tres pacientes de sexo feminino en las que las dos entidades se presentaron en forma simultánea. Sugerimos la utilización de inmunosupresores junto con tratamiento de plasmaféresis con reposición de sobrenadante de crioprecipitado. (AU)DEFINICIÓN:
L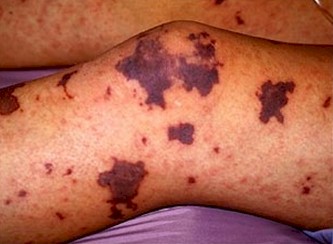 a púrpura trombótica trombocitopénica es una vasculitis generalizada que afecta al endotelio de arteriolas y capilares de muchos órganos, en particular del cerebro, corazón, hígado, páncreas y riñón. Es considerada como una emergencia médica que puede ser potencialmente fatal. Se clasifica dentro de las microangiopatías trombóticas, un término anatomopatológico que describe la existencia de trombosis en los pequeños vasos del organismo. Algunos autores adoptan esta denominación para referirse en su conjunto a dos enfermedades: el síndrome urémico-hemolítico (SUH) y la púrpura trombótica trombocitopénica (PTT), los cuales comparten muchas características comunes.
a púrpura trombótica trombocitopénica es una vasculitis generalizada que afecta al endotelio de arteriolas y capilares de muchos órganos, en particular del cerebro, corazón, hígado, páncreas y riñón. Es considerada como una emergencia médica que puede ser potencialmente fatal. Se clasifica dentro de las microangiopatías trombóticas, un término anatomopatológico que describe la existencia de trombosis en los pequeños vasos del organismo. Algunos autores adoptan esta denominación para referirse en su conjunto a dos enfermedades: el síndrome urémico-hemolítico (SUH) y la púrpura trombótica trombocitopénica (PTT), los cuales comparten muchas características comunes.
La púrpura trombótica trombocitopénica es más frecuente en las mujeres que en los hombres y el pico de incidencia se sitúa entre los 30 y 40 años. Clínicamente, se caracteriza por 5 signos clave, descritos inicialmente por Moschcowitz en 1924:
* Anemia hemolítica
* trombocitopenia
* neuropatía
* fiebre
* insuficiencia renal
Hasta la década de los 80, la púrpura trombótica trombocitopénica era fatal en la mayoría de los casos. Con la introducción de las transfusiones de intercambio de plasma, hoy día es una enfermedad que puede ser tratada de forma efectiva en el 80-90% de los casos.
era fatal en la mayoría de los casos. Con la introducción de las transfusiones de intercambio de plasma, hoy día es una enfermedad que puede ser tratada de forma efectiva en el 80-90% de los casos.
Se conocen varios tipos de PTT. La PPT crónica recurrente, usualmente congénita, se manifiesta en la infancia con recurrencias a intervalos de 3 semanas de forma indefinida. Estas recurrencias son predecibles y pueden ser prevenidas mediante infusiones de plasma.
La PTT idiopática es la forma más frecuente y más grave de PTT. Se manifiesta entre el final de la adolescencia y la edad madura y un 60% de los pacientes tratados con éxito no vuelven a recurrir. En el resto, la enfermedad recurre de forma intermitente y no predecible.
a púrpura trombótica trombocitopénica es una vasculitis generalizada que afecta al endotelio de arteriolas y capilares de muchos órganos, en particular del cerebro, corazón, hígado, páncreas y riñón. Es considerada como una emergencia médica que puede ser potencialmente fatal. Se clasifica dentro de las microangiopatías trombóticas, un término anatomopatológico que describe la existencia de trombosis en los pequeños vasos del organismo. Algunos autores adoptan esta denominación para referirse en su conjunto a dos enfermedades: el síndrome urémico-hemolítico (SUH) y la púrpura trombótica trombocitopénica (PTT), los cuales comparten muchas características comunes.
La púrpura trombótica trombocitopénica es más frecuente en las mujeres que en los hombres y el pico de incidencia se sitúa entre los 30 y 40 años. Clínicamente, se caracteriza por 5 signos clave, descritos inicialmente por Moschcowitz en 1924:
* Anemia hemolítica
* trombocitopenia
* neuropatía
* fiebre
* insuficiencia renal
Hasta la década de los 80, la púrpura trombótica trombocitopénica
era fatal en la mayoría de los casos. Con la introducción de las transfusiones de intercambio de plasma, hoy día es una enfermedad que puede ser tratada de forma efectiva en el 80-90% de los casos.
Se conocen varios tipos de PTT. La PPT crónica recurrente, usualmente congénita, se manifiesta en la infancia con recurrencias a intervalos de 3 semanas de forma indefinida. Estas recurrencias son predecibles y pueden ser prevenidas mediante infusiones de plasma.
La PTT idiopática es la forma más frecuente y más grave de PTT. Se manifiesta entre el final de la adolescencia y la edad madura y un 60% de los pacientes tratados con éxito no vuelven a recurrir. En el resto, la enfermedad recurre de forma intermitente y no predecible.
Esta enfermedad puede ser causada por la falta de una enzima (un tipo de proteína) que está involucrada en la coagulación de sangre. El hecho de no tener suficiente cantidad de esta enzima provoca la coagulación. A medida que las plaquetas se agrupan en estos coágulos, hay menos cantidad de ellas disponibles en la sangre en otras partes del cuerpo para ayudar con la coagulación. Esto puede llevar a sangrado bajo la piel y manchas de color violeta llamadas púrpura.
SÍNTOMAS:
* Sangrado en la piel o membranas mucosas
* Cambios en el estado de conciencia
* Confusión
* Fatiga fácil
* Fiebre
* Dolor de cabeza
* Frecuencia cardíaca sobre 100 latidos por minuto
* Palidez
* Manchas purpurinas en la piel producidas por pequeños vasos sangrantes cerca de la superficie cutánea (púrpura)
* Dificultad respiratoria al esforzarse
* Cambios en el habla
* Debilidad
* Color amarillento de la piel (ictericia)
* Cambios en el estado de conciencia
* Confusión
* Fatiga fácil
* Fiebre
* Dolor de cabeza
* Frecuencia cardíaca sobre 100 latidos por minuto
* Palidez
* Manchas purpurinas en la piel producidas por pequeños vasos sangrantes cerca de la superficie cutánea (púrpura)
* Dificultad respiratoria al esforzarse
* Cambios en el habla
* Debilidad
* Color amarillento de la piel (ictericia)
DIAGNÓSTICO:
El diagnóstico de la PPT se basa en el comportamiento clínico de la enfermedad ya que la presentación de todos los tipos es muy similar. La mayoría de los pacientes muestan una acusada trombocitopebia, anemia hemolítica debida a la fragmentación de los eritrocitos (esquistocitosis), niveles elevados de la LDH, signos de isquemia y síntomas relacionados con el sistema nervioso central (alteraciones de la memoria, comportamiento irregular, cefaleas, etc). Los dos signos biológicos de mayor valor diagnóstico son la morfologia eritrocitaria (hematíes fragmentados, esquistocitos, queratocitos y microesferocitos) y una intensa trombopenia (<>
El diagnóstico de la PPT se basa en el comportamiento clínico de la enfermedad ya que la presentación de todos los tipos es muy similar. La mayoría de los pacientes muestan una acusada trombocitopebia, anemia hemolítica debida a la fragmentación de los eritrocitos (esquistocitosis), niveles elevados de la LDH, signos de isquemia y síntomas relacionados con el sistema nervioso central (alteraciones de la memoria, comportamiento irregular, cefaleas, etc). Los dos signos biológicos de mayor valor diagnóstico son la morfologia eritrocitaria (hematíes fragmentados, esquistocitos, queratocitos y microesferocitos) y una intensa trombopenia (<>